
FLAM-Seq: Genome-wide full-length mRNA and poly(A)-tail sequencing
Keywords
mRNA,NGS,poly(A)-tail,RNA biology,sequencing
Invention Novelty
Detailed information about mRNA isoform expression including respective poly(A)-tail sequence (actual sequence length and sequence composition) is invaluable for the in-depth understanding of cellular gene regulation and RNA biology. The proposed invention provides an innovative solution for cDNA library preparation, enabling transcriptome-wide sequencing of full-length mRNAs including respective poly(A)-tails.
Value Proposition
While NGS-based technologies allow analysis of alternative polyadenylation sites determining the 3ʹ-UTR length of gene transcripts, poly(A) length and composition demonstrating dynamic and developmental regulation remain poorly investigated. Current assay systems attain only long-read sequences or poly(A) length estimation in a separate manner, or simultaneously produce long reads and quantify poly(A) tails, limited by high material input and low accuracy. Hence, it remains difficult to acquire comprehensive information of the entire “mRNA architecture” (assembled coding regions, non-coding regions and corresponding poly(A)-tail) at once for a given gene on a quantitative level in a genome-wide transcriptomics approach. With its outstanding efficiency and accuracy, the here proposed Full-length poly(A) mRNA sequencing (FLAM-Seq) technology offers the potential to unravel important aspects of RNA biology.
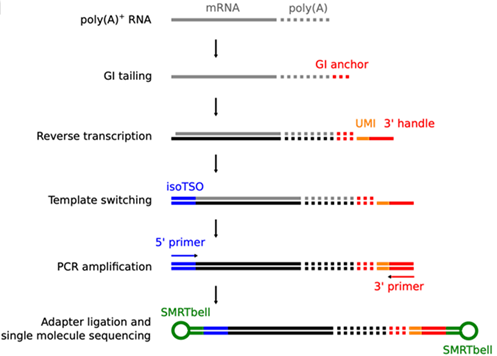
Underlying principle for Full-length poly(A) and mRNA sequencing: FLAM-seq (Legnini et al., 2019).
Technology Description
FLAM-Seq comprises a versatile protocol, enabling the annotation of mRNA isoforms and their abundance in conjunction with respective poly(A)-tail length. Instead of poly(A)-length estimation, FLAM-Seq reveals the global distribution of poly(A)-length and ultimately uncovers alternative nucleotide compositions within the poly(A)-sequence. No sequencing bias occurs when determining long Adenosine-sequence stretches. Sequencing of the “Full-length”-library based on this method guarantees high reproducibility, with long comprehensive reads. The researchers successfully validated the method in a wide variety of tissues - human cancer cells, healthy and Herpes-infected brain organoids, nematode cells, flies, mouse brains and octopus. This ubiquitous applicability and its outstanding accuracy qualify FLAM-Seq as highly valuable tool for e.g., taxonomic studies and whole-genome annotations.
Commercial Opportunity
This opportunity is available for in-licensing.
Development Status
Protocols for the methodology have been established, optimized, and validated.
Patent Situation
Patents pending in Europe (EP3861133A1), US (US2022002797A1) and Canada (CA3112262A1) with priority of 2018.
Further Reading
Legnini, I., Alles, J., Karaiskos, N. et al. FLAM-seq: full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat Methods 16, 879–886 (2019).
Licensing Contact

Technology Scout
T: +49 30 450 543078
christiansen@ascenion.de
Institute of Origin
